
Human Genetics | Single-cell Biology | Multi-omics | Modifiers
Can therapeutic interventions be used to reduce deleterious effects?
I develop and utilize statistical and computational approaches across population biobanks and single-cell genomics to address these questions, collaborating closely with experimental labs, clinicians, and large consortia to validate and test mechanistic and therapeutic hypotheses.
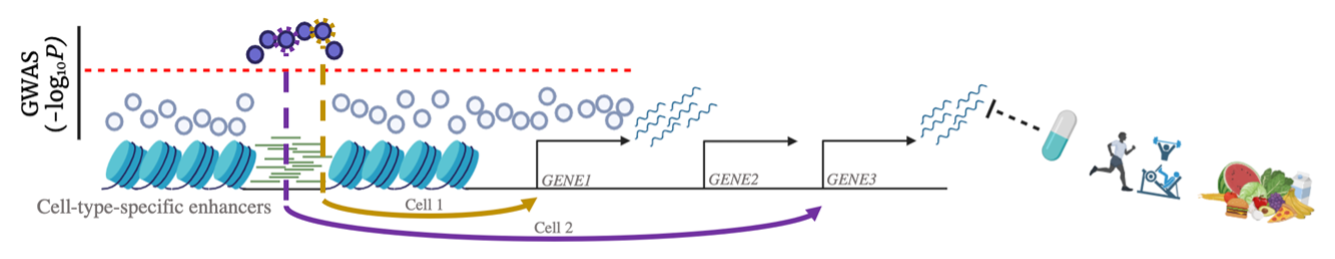
1. How do complex genetic risk factors (involving multiple genes) drive high rates of disease?
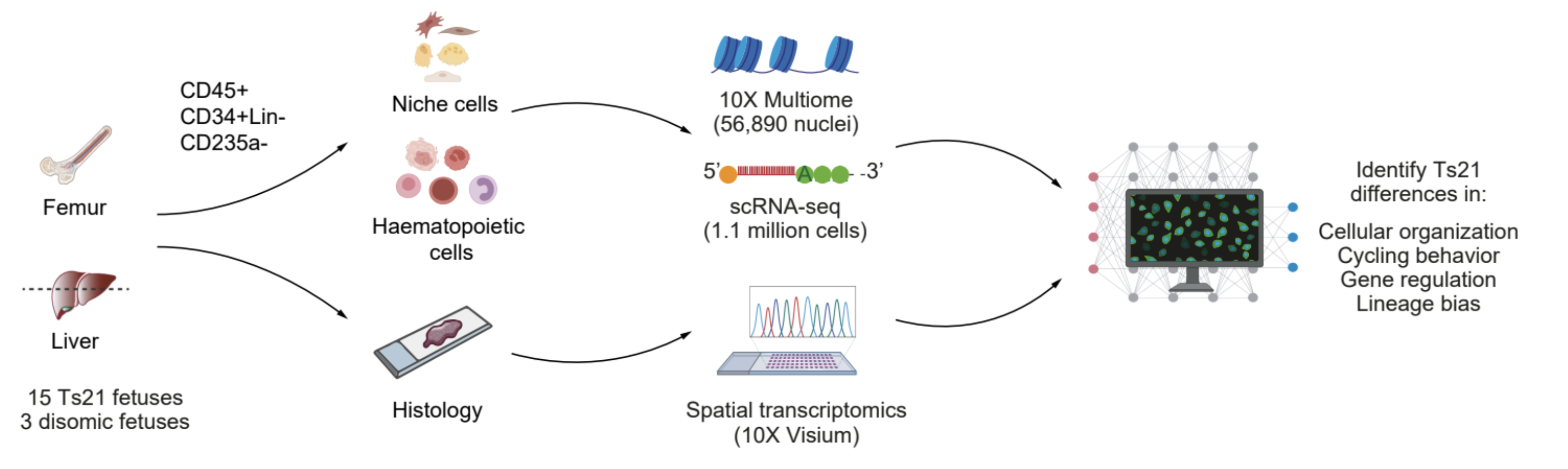
While individual variants and genes influence disease risk, it is unclear how they collectively shape risk. Single-cell multi-omics is a powerful technology for studying complex genetic risk factors. My postdoc research focused on fetal Down Syndrome (trisomy 21 or Ts21), which is a unique genetic model because of a large molecular impact (fetal hematopoiesis is defective, driving 150-fold higher risk for childhood leukemia and red blood cell (RBC) overproduction) stemming from an already-identified cellular context (fetal hematopoietic stem cells (HSCs)) and known genetic “variant” (Ts21). While Ts21 leukemogenesis is initiated before birth, the molecular changes that occur prior to key somatic mutation events are unclear. In collaboration with Dr. Stephen Montgomery’s and Dr. Ana Cvejic’s labs, I led a study using 1.1m scRNA-seq cells, 10X multiome, and spatial transcriptomics from fetal liver and femur. Our analysis found that gene expression differences were cell-type- and environment-specific; and by leveraging GWAS SNPs, we unveiled modified enhancer-gene maps that bias Ts21 HSCs towards the erythroid lineage. Notably, our results point to a key hypothesis where altered chromatin accessibility and heightened oxidative stress underlie increased mutational burden near leukemia-related genes. We are actively validating our findings and testing our hypotheses, exposing a novel perspective on leukemia etiology in Ts21 (Marderstein et al. In press at Nature).
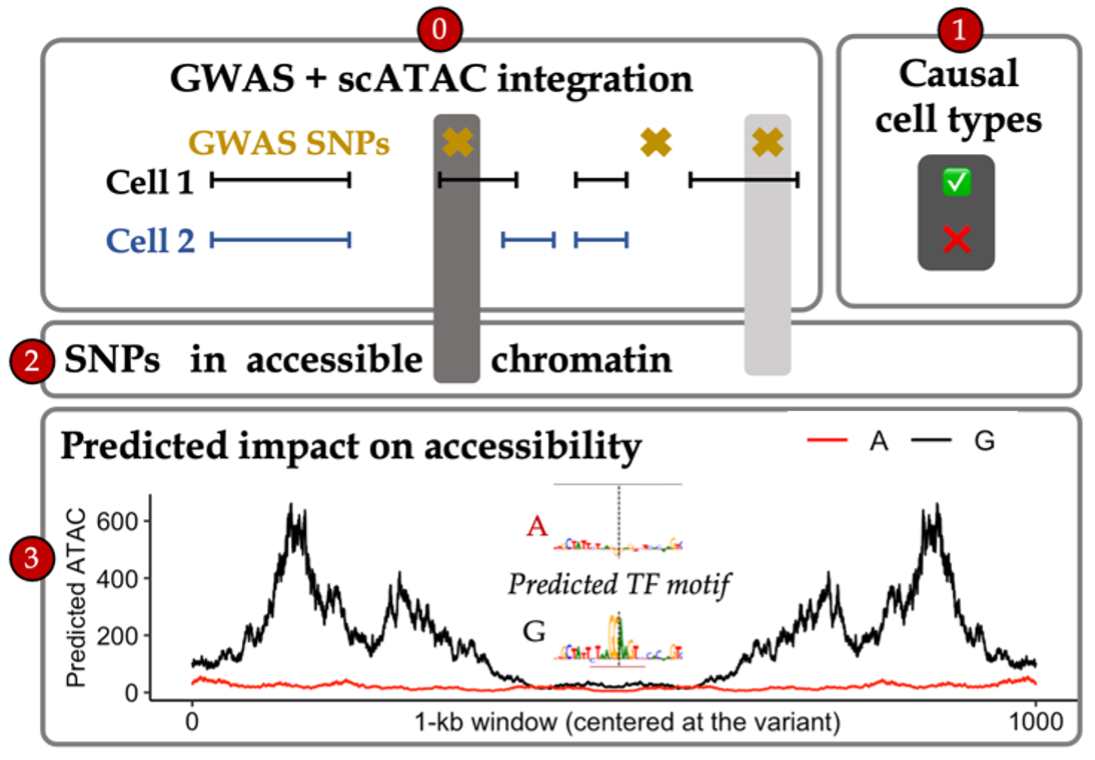
2. Which variants are causal and how do they exert their effects in disease-relevant cells?
The majority of GWAS loci reside in non-coding regions. However, translating non-coding associations into mechanistic insights of disease is limited by incomplete knowledge of: (1) causal variants (due to linkage disequilibrium), (2) relevant cellular contexts, and (3) regulatory mechanisms linking SNPs to genes (which are cell-type-specific).
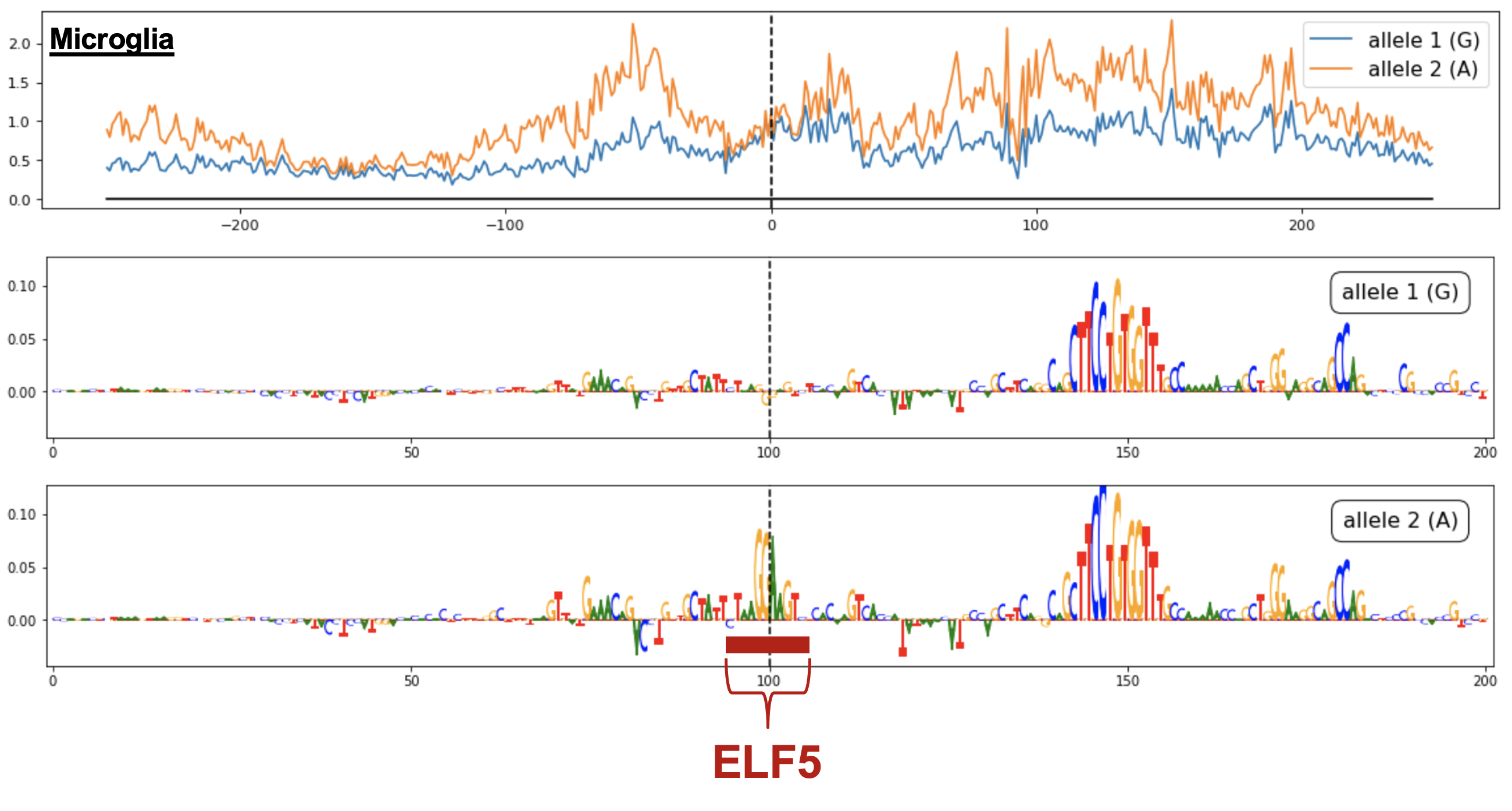
I developed a framework in collaboration with Soumya Kundu from Dr. Anshul Kundaje’s group for understanding how variants act within different cellular contexts. Broadly, using scATAC-seq, we (i) establish a single-cell map of causal cell types (by assessing GWAS variant enrichment in accessible chromatin), (ii) identify non-coding GWAS variants which reside in accessible chromatin of disease-relevant cell types, and (iii) predict the cell-type-specific effects of prioritized variants by training convolutional neural networks (called chromBPnet) that learn regulatory syntax and predict scATAC-seq profiles from DNA sequence.
By applying our framework across neurological diseases using scATAC-seq cells from fetal and adult brain samples, we prioritized causal variants acting through causal cell types across traits (e.g. multiple microglia-specific variant effects at the non-coding Alzheimer’s locus near PICALM) and partitioned new disease pathways acting through individual cell types (e.g. ion transport regulation in Alzheimer’s via microglia). Furthermore, as our models can be applied to any base pair in the genome, we predicted regulatory effects at 10 million rare variants found in the human population. While the vast majority of these have negligible effects, our predictions helped prioritize non-coding rare variants involved in common disease and infer the selective pressures acting on regulatory variation. This demonstrates the potential to build context-specific regulatory maps which interpret the effects of non-coding loci in particular cell populations for multiple diseases (Marderstein et al. ASHG Mtg 2022, 2024 Presentations).
As our approaches can be applied across diseases, I have been expanding our approaches in the IGVF Consortium. I led the development of a multi-ancestry functional genomics resource with >40 collaborators to prioritize systemic lupus erythematosus (SLE) mechanisms. Collaborating in the IGVF Consortium has allowed us to integrate multiple data modalities for understanding variant impacts on genome function and disease. We (i) illuminated population-specific SLE variation, (ii) predicted regulatory consequences of non-coding SLE variants across primary immune cells (particularly B cells), and (iii) prioritized thousands of variants for CRISPR base editing and MPRAs (Marderstein et al. IGVF Mtg 2023 Presentation). As SLE has varying prevalence among ancestries, this work lays down the foundation to distinguish mechanisms contributing to health disparities.

3. How does environment modulate genetic effects, and can we use this information for therapeutic benefit?
Specialized wearable tracking devices and improvements in biomarker data are being explored, and the hope is that these will deliver a quantum improvement in the availability and accuracy of environmental data. However, gene-environment (GxE) interaction testing suffers from a dimensionality problem when testing for the numerous possible interactions between genome-wide variants and many environmental factors. As a result, efficiently identifying GxE interactions is an important statistical and computational challenge.
I first published a new statistical method to identify variants influencing variability in traits (vQTLs), which were enriched for GxE and operate through pathways and cell types both akin to and distinct from the conventional GWAS loci (Marderstein et al. 2021a AJHG).

Our success led to the realization that environment modulates variant effects similarly across those involved in the same pathway, such that the impact of variants is consistently amplified in some key contexts. We recently found this amplification to be pervasive across environments and phenotypes (Poyraz, Clark, & Marderstein ASHG Mtg 2022 Presentation, SMBE Mtg 2021 Presentation).
Notably, as commonly-used medications present a promising set of modifiable factors to reduce polygenic risk, I tested for interactions between breast cancer risk and medication usage. I found that corticosteroid use exacerbates polygenic risk of breast cancer through NRF2-mediated gene regulation (Marderstein et al. 2021b AJHG). This demonstrated an exciting opportunity for envisioning GxE in precision medicine, such as identifying personalized strategies for treating individuals with high polygenic risk.

Interested in collaborating? Contact me to discuss potential projects or partnerships.