
Research Program
From single-nucleotide variants to whole-chromosome aneuploidies, a central challenge in biology is understanding how genetic alterations reshape gene regulation across development, aging, and cancer. The widespread adoption of whole-genome sequencing (WGS), from newborn screening and population health to somatic sequencing of healthy and cancerous tissues, has generated an unprecedented catalog of genetic variation. However, our ability to sequence genomes has far surpassed our ability to understand them. Billions of inherited and somatic variants have shifted the challenge from generating data to interpreting how they disrupt gene regulatory networks, alter cellular identity, and create lineage-specific susceptibilities to disease. My research seeks to understand how genetic alterations reshape gene regulation across the lifespan by integrating population genetics, single-cell genomics, machine learning, and functional genomics.
1. How does aneuploidy reshape cellular development, ultimately initiating cancer development?
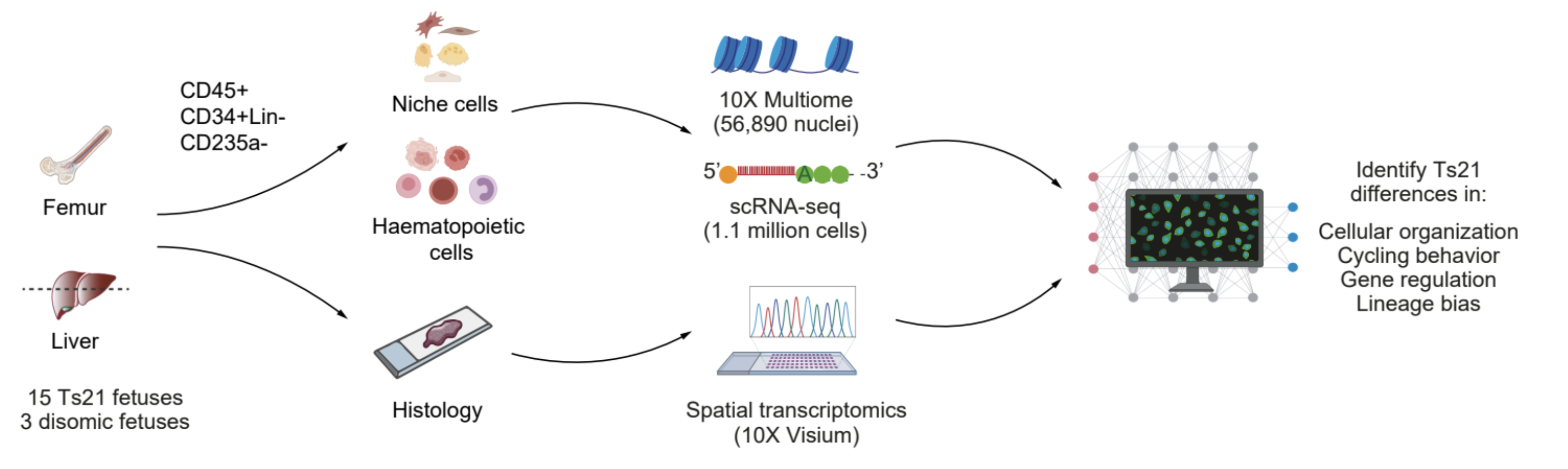
Children with Down Syndrome (DS, trisomy 21) face 150-fold higher risk of leukemia by age 5, with profound hematological abnormalities before cancer occurs. However, while the initial cancer mutations appear before birth and are unique to Down Syndrome, it is unclear why they appear in the first place. In collaboration with Dr. Stephen Montgomery’s and Dr. Ana Cvejic’s labs, using multimodal data spanning scRNA-seq, 10x multiome, and nano-CUT&Tag across >1 million cells with Hi-C and Visium spatial profiling, I mapped the human fetal hematopoietic system during this early pre-leukemic window in Down Syndrome and Edwards Syndrome (trisomy 18). In DS, I found that trisomy 21 induces oxidative stress, rewires enhancer-gene interactions, and influences chromatin organization to alter hematopoiesis: a novel finding on leukemia etiology in Down Syndrome that I published in Nature. More recently, using deep learning models of trisomy 18 and trisomy 21 cells to interpret TF networks, these data revealed a unifying model describing how aneuploidy reorganizes developmental gene regulatory networks to alter cell fate (manuscript unpublished/in revision).
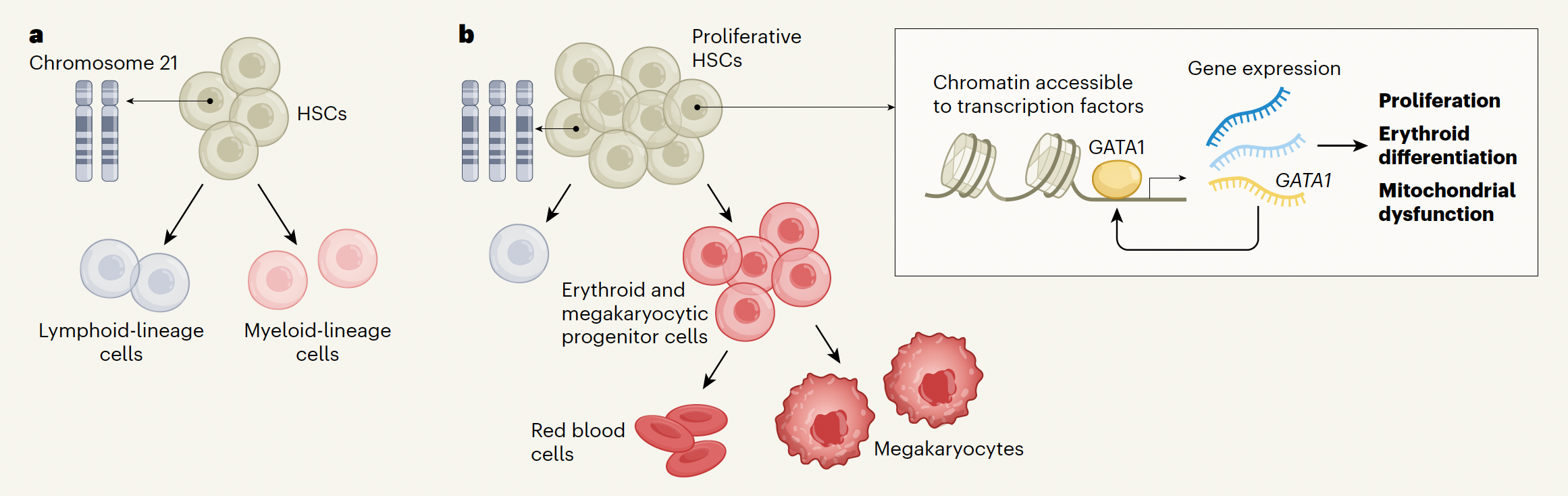
In cancer, aneuploidy is one of the most common events, yet its effects on gene regulation and cell state are poorly understood. In tumors, aneuploidy emerges alongside many other molecular changes, making it difficult to distinguish their consequences. Therefore, germline trisomies provide a natural human model of chromosome dosage imbalance and a blueprint for understanding the consequences of somatic aneuploidy by tracing their effects through the earliest stages of development. My recent findings suggest that regulatory programs revealed by germline aneuploidy can re-emerge in cancer, providing a framework to discover novel oncogenic mechanisms and therapeutic strategies shaped by somatic chromosome dosage. My work seeks to develop a general framework for interpreting somatic chromosome dosages by integrating clinicogenomic data, single-cell multi-omics, and functional perturbation, and leveraging insights from germline aneuploidies to understand how regulatory circuits are rewired in cancer.
2. How do non-coding genetic factors influence disease through cell-type-specific regulation?
Over two decades of GWAS have established non-coding regions as a significant contributor to human disease, as the majority of GWAS loci reside in non-coding regions. However, translating non-coding associations into mechanistic insights of disease is limited by incomplete knowledge of: (1) causal variants (due to linkage disequilibrium), (2) relevant cellular contexts, and (3) regulatory mechanisms linking SNPs to genes (which are cell-type-specific). To address this, I have developed methods that integrate deep learning, population genetics, and functional genomics to systematically interpret regulatory variation.
In collaboration with Soumya Kundu from Dr. Anshul Kundaje’s group, we used a framework to predict the cell-type-specific variant effects by training sequence-to-function deep learning models that learn regulatory syntax and predict scATAC-seq profiles from DNA sequence. As our models could be applied to any base pair in the genome, Soumya and I generated a resource of 3 billion variant-by-context predictions across >100 fetal and adult cellular contexts, aiding interpretation of disease variants and guiding experimental follow-up.
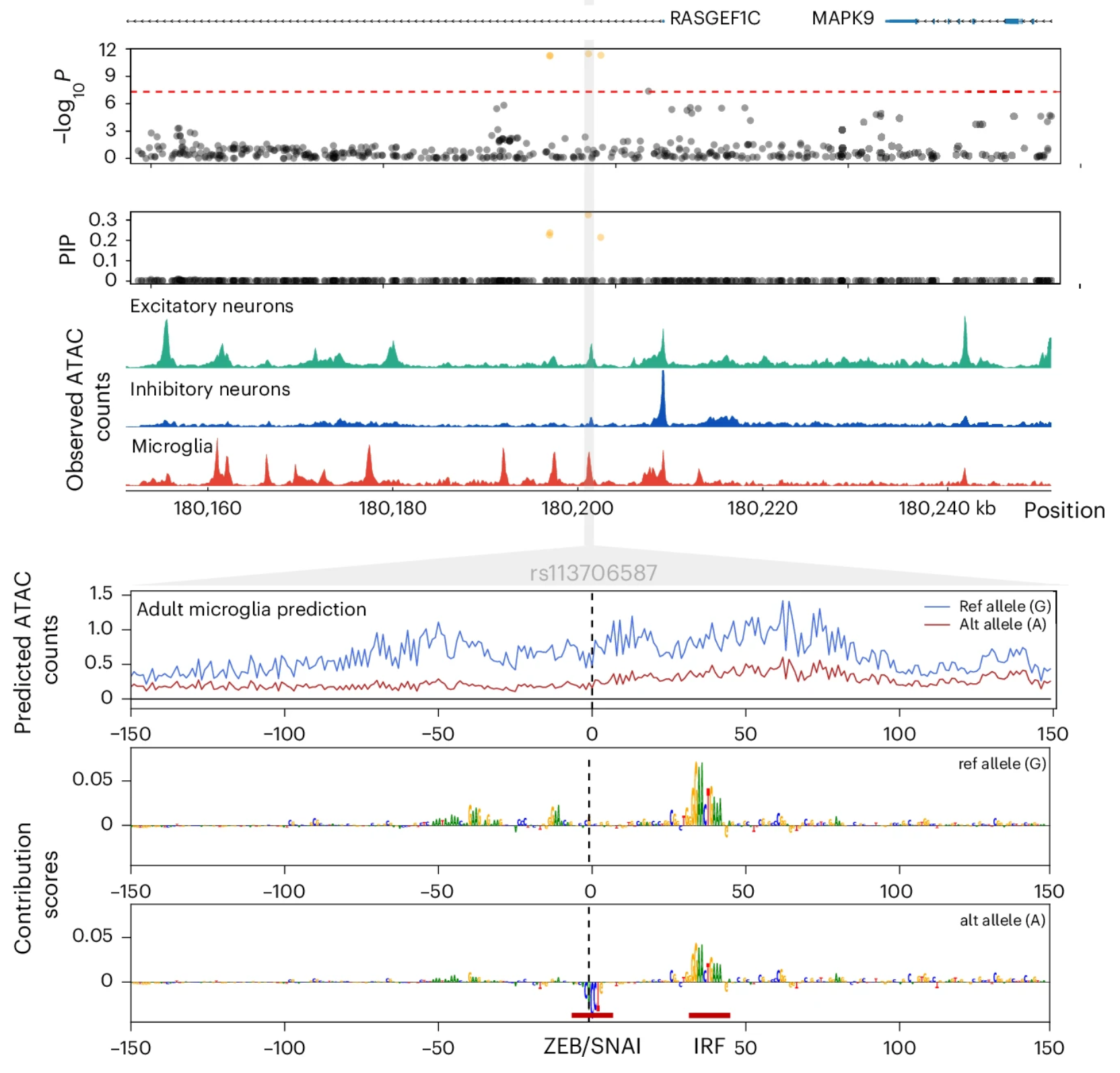
Importantly, this revealed fundamental differences between common and rare variants, where rare variants are more likely to have broad regulatory effects across multiple cell types while common variants are more cell-type-specific. This led to developing FLARE, which integrates evolutionary constraint to prioritize non-coding variants with extreme functional effects. Applying FLARE to cohorts of families affected by autism spectrum disorder or congenital heart disease identified de novo mutations enriched in affected individuals but largely absent from controls, demonstrating the power of integrating population genetic signatures to interpret rare non-coding variation. We published FLARE in Nature Genetics.
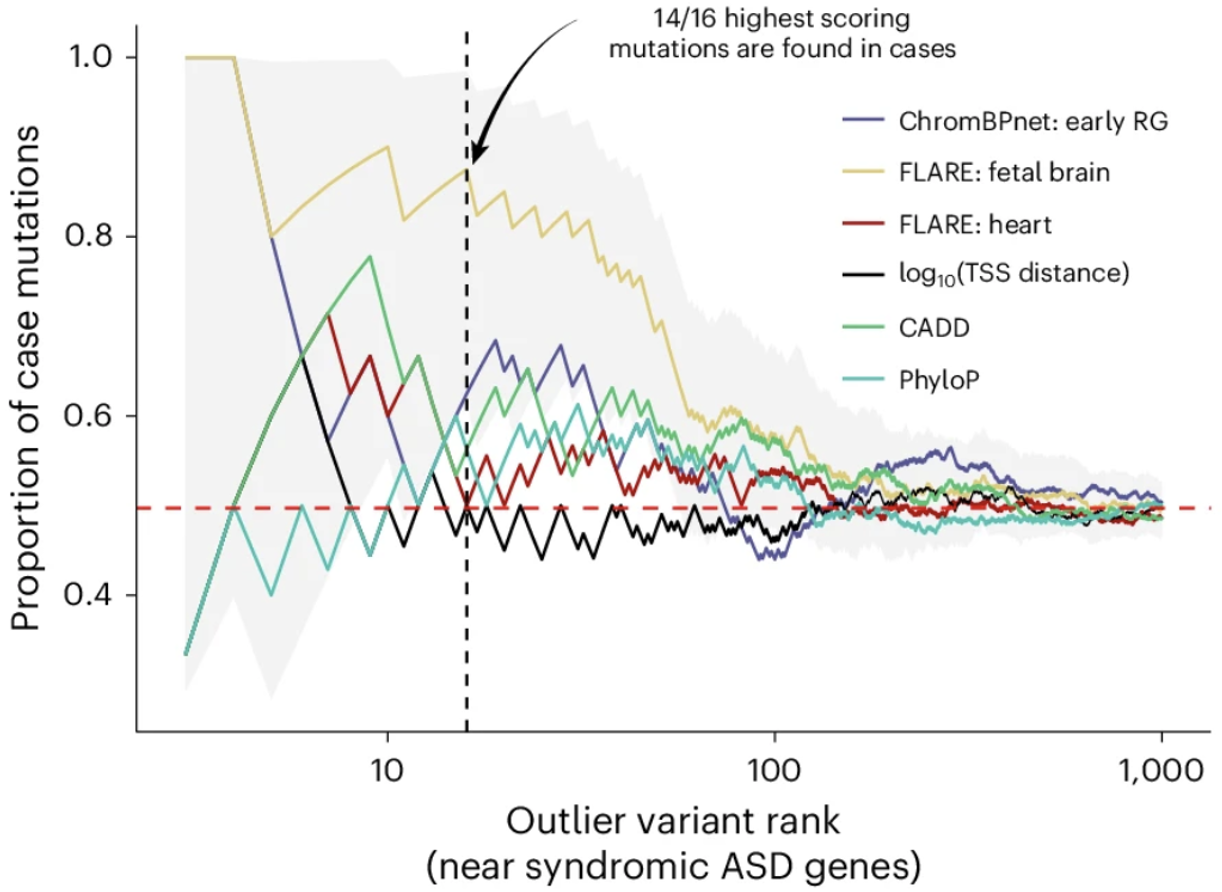
At Memorial Sloan Kettering, I am actively working on approaches to more directly interpret the regulatory mechanisms learned by deep learning models using feature attribution methods. I am applying these methods to interpret clinical mutations, establishing a framework for linking non-coding variation to clinical and mechanistic interpretations of regulatory variation.
As our approaches can be applied across diseases, I have been also applied these approaches in the IGVF Consortium. I led the development of a multi-ancestry functional genomics resource with >40 collaborators to prioritize thousands of systemic lupus erythematosus (SLE) variants and their mechanisms for base editing and massively-parallel reporter assay experiments. As SLE has varying prevalence among ancestries, this work illuminated significant population-specific variation and laid down the foundation to distinguish mechanisms contributing to health disparities.

3. How does environment modulate genetic effects, and can we use this information for therapeutic benefit?
Specialized wearable tracking devices, satellite imaging, and molecular biomarker data are being developed, and the hope is that these will deliver a quantum improvement in the availability and accuracy of environmental data. However, gene-environment (GxE) interaction testing suffers from a dimensionality problem when testing for the numerous possible interactions between genome-wide variants and many environmental factors. As a result, efficiently identifying GxE interactions is an important statistical and computational challenge.
I developed the Deviation Regression Model to improve GxE discovery by first identifying variants associated with variability (vQTLs). The Deviation Regression Model remains one of the best-performing approaches for vQTL detection. Furthermore, these vQTLs were enriched for GxE interactions and operate through pathways and cell types both akin to and distinct from the conventional GWAS loci (Marderstein et al. 2021a AJHG), providing a new window into the genetic basis of complex phenotypes.

Our success led to the realization that environment modulates variant effects similarly across those involved in the same pathway, such that the impact of variants is consistently amplified in some key contexts. We recently found this amplification to be pervasive across environments and phenotypes (Poyraz, Clark, & Marderstein ASHG Mtg 2022 Presentation, SMBE Mtg 2021 Presentation).
Notably, as commonly-used medications present a promising set of modifiable factors to reduce polygenic risk, I tested for interactions between breast cancer risk and medication usage. I found that corticosteroid use exacerbates polygenic risk of breast cancer through NRF2-mediated gene regulation (Marderstein et al. 2021b AJHG). This demonstrated an exciting opportunity for envisioning GxE in precision medicine, such as identifying personalized strategies for treating individuals with high polygenic risk.

Interested in collaborating? Contact me to discuss potential projects or partnerships.